Waqar A Khan
Department of Biochemistry and Molecular Biology, Dhaka Shishu Hospital, Sher-e-Bangla Nagar, Dhaka, Bangladesh.
Bilquis Banu
Department of Clinical Pathology, Dhaka Shishu Hospital, Sher-e-Bangla Nagar, Dhaka, Bangladesh.
Md. Abdul Aziz
Department of Biochemistry and Molecular Biology, Dhaka Shishu Hospital, Sher-e-Bangla Nagar, Dhaka, Bangladesh.
Salma Sadiya
Department of Biochemistry and Molecular Biology, Dhaka Shishu Hospital, Sher-e-Bangla Nagar, Dhaka, Bangladesh.
Md. Belayet Hossain
Department of Haematology and Oncology, Dhaka Shishu Hospital, Sher-e-Bangla Nagar, Dhaka, Bangladesh.
Md Selimuzzaman
Department of Haematology and Oncology, Dhaka Shishu Hospital, Sher-e-Bangla Nagar, Dhaka, Bangladesh.
Keywords: Beta Thalassemia mutations Xmn1 Polymorphism, Restriction enzyme, Heterozygous, Homozygous.
DOI: 10.3329/bmrcb.v47i2.57783
Abstract
Background: In Bangladesh, more than 14000 children on are born annually with in thalassaemia – a common congenital disease Hb E trait is 6.1%. Hb E beta thalassaemia is the most common type of thalassaemia, followed by Beta thalassaemia major.
Objectives: To determine the frequency of Xmn1 polymorphism and its association with Beta thalassaemia mutations.
Methods: A total of one hundred and four Bangladeshi thalassaemia patients were analysed. Amplification Refractory Mutation System (ARMS) was utilized for Beta thalassaemia mutations and digestion of the PCR product using Xmn1 restriction enzyme Pdml for Xmn1 polymorphism.
Results: Xmn1 polymorphism was detected in seventy patients of which 60(57.69%) were heterozygous for Xmn1 polymorphism and seventeen (16.35%) were homozygous. The most common genotype found was heterozygous Xmn1(-/+)seen in 57.70%. The age of presentation of thalassaemic patients was delayed in those who had Xm1 polymorphism.The mean age of presentation of Hb E beta thalassaemia was 13.35 years having homozygous Xm1 polymorphism,7.21 years in heterozygous and 6.25 years without Xmn 1 polymorphism. The most common mutation detected was Cd26 (G-A) +IVS 1-5(G-C) in fifty eight patients in which thirty nine (67.24%) were heterozygous for Xmn 1 polymorphism and 8 (13.79%) were homozygous (+/+).The second most common mutation observed was Cd26(G-A)+30(G-C) seen in fourteen patients where 57.14% were homozygous for Xmn 1 polymorphism and 35.71% were heterozygous. In thalassaemia major 9 (90%) were negative for Xmn1 polymorphism. Allele frequency of Xmn 1 polymorphism was 0.45.
Conclusion: The association of Xmn1 polymorphism with two common mutations seen in Hb E beta thalassemia patients may be utilized for hydroxyurea therapy to reduce the requirement of blood transfusion.
Keywords: Beta Thalassemia mutations Xmn1 Polymorphism, Restriction enzyme, Heterozygous, Homozygous.
Introduction
Thalassaemia is a very common congenital disorder in Bangladesh.It is estimated that more than fourteen thousand thalassaemia children are born annually. The carrier frequency of beta thalassaemia is 4.1% and Hb E trait is 6.1%.1,2 Hb E beta thalassaemia is the most common type of thalassaemia seen followed by Beta thalassaemia major.3-5
Most of our patients cannot afford any treatment or have inadequate treatment , but somehow manage to have a blood transfusion but not enough to maintain Hb level as recommended. For many years efforts are being made to reduce the globin chain imbalance in patients with thalassaemia and sickle cell anaemia by augmenting the fetal haemoglobin synthesis by drugs like 5-azacytidine,butric acid recombinant human erythropoiten and hydroxyurea (HU) or with combinations of these drugs.6,7
The presence of the Xmn1 polymorphism in the Gy globin gene promoter has been shown to improve the severity of the disease due to increase production of Hb F in conditions of erythropoietic stress.8-10 Presence of Xmn1 polymorphism has also shown to reduce the requirement of blood transfusion in thalassaemia patients or even lead to cessation of blood transfusion as a result of therapy by hydroxyurea as reported in some studies.11-14
The frequency of Xmn1 polymorphism varies in different geographical regions of the world.15-18 It also has been reported to correlate with certain beta thalassaemia mutations.11,12,17,19,20 Xmn 1 polymorphism association with HbE mutation has also been observed.21
This study was aimed to determine the frequency of Xmn1 polymorphism and its correlation with beta thalassaemia mutations in our patients.This is the first study regarding the frequency of Xmn1 polymorphism, and its correlation with Beta thalassaemia mutations seen in Bangladeshi thalassaemic patients.
Materials and Methods
The study was done in the DNA lab of the Department of Biochemistry and Molecular Biology, Dhaka Shsihu(Children) Hospital, Dhaka, Bangladesh. 104 thalassaemia patients were randomly selected , who came to Dhaka Shishu Hospital Thalassemia Center. The patients were analysed for Beta Thalassemia mutations and Xmn1 polymorphism. Ages of patients at the time of diagnosis were recorded. There were 85(81.73%) Hb E beta Thalassaemia patients and 19(18.27%) Beta thalassaemia major patients.Fifty seven (54.81%) were males and forty seven(45.19%) were females. The period of study was for six months from January, 2018 to July, 2018.
2 cc of blood was collected in EDTA containing tubes. DNA extraction was done by Purelink Genomic DNA mini kit (Invitogen, USA).Mutations in the beta globin chain was analyzed by allele specific primers using Amplification Refractory Mutation System-Polymerase Chain Reaction(ARMS-PCR). Extracted DNA samples were stored at -200 C for detection of Xmn 1 polymorphism.
Detection of Xmn 1 polymorphism
Detection of GãXmnI polymorphism of C to T base pair substitution at the “158 position in the promoter region of the Gã-globin gene (“158 (C > T) Xmn1 polymorphism): The Polymerase chain reaction (PCR) was done by Taq polymerase, DNTP, template, MgCl and Xmn1 primer ( 52 -AAC TGT TGC TTT ATA GGA TTT T-32 and 52 - AGG AGC TTA TTG ATAACT CAG AC-32 .
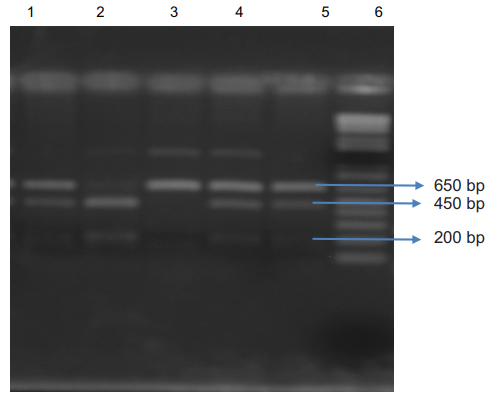
The cycling reaction was performed under the following conditions: Denaturation at 95 °C for 10 min, 30 cycles of denaturation at 94°C for 1 min, primer annealing at 55°C for 1 min, extension at 72°C for 1 min and final extension at 72 °C for 10 min. The PCR products were digested with the Xmn1 Restriction enzyme (Pdml) for 6-8 hours. Electrophoresis was done on these digested products on a 2% agarose gel. Amplification with the primers produced a 650 bp fragment in a patient with Xmn 1 (-/-) genotype,650,450 bp and 200 bp fragments from a patient with heterozygote for Xmn 1 (+/-) genotype and 450 bp and 200 bp fragments from a patient with Xmn1 (+/+) genotype Fig 1 Written consent was taken from each patient/guardian. The study was approved by the ethical committee of Bangladesh Medical and Research Council. (Figure 1).
Lane 1, 4, 5: 650 bp, 450 bp and 200 bp fragments from patients heterozygous for the Xmn I (+/-) genotype; Lane 2: 450 bp and 200 bp digested fragments from a patient with Xmn I (+/+) genotype; Lane 3: 650 bp fragment from a patient with Xmn1 (-/-) genotype Lanes 6: 1kb DNA ladder
Results
A total of 104 thalassemia patients were analysed for Xmn1 polymorphism and Beta thalassemia mutations. There were 85 (81.73%) Hb E beta thalassaemia and 19 (18.27%) Beta thalassemia major patients. Fifty seven (54.81%) were males and forty seven (45.19%) were females. The mean age at the time of diagnosis of Hb E beta thalassaemia patients was 13.35 years in Xmn1(+/+) ,7.21 years in Xmn1(-/+) and 6.25 years in Xmn1 (-/-). In thalassaemia major patients the mean age of the presentation was 5 years in Xmn1(-/+) and 2.76 years in Xmn1(-/-) (table I).
Types of Thalassaemia |
Xmn1 (-/-) |
Xmn1 (-/+) |
Xmn1 (+/+) |
|---|---|---|---|
Beta thalassaemia major |
2.76 |
5.0 |
0 |
Hb E beta thalassaemia |
6.25 |
7.21 |
13.35 |
Eight beta globin gene mutations were detected in âo thalassemia. Beta thalassaemia mutations detected were IVS 1-5(G-C) HBB:c.92+5G>C, 30(G-C) HBB:c.91G>C, 130(G-C) HBB:c.93-1G>A,15(G-A) HBB:c.47G>A, 30(G-A) HBB:c.92G>A, Fr 42-43 HBB:c.126_129delCTTT, Fr 8-9 HBB:c.27_28insG, 15(-T) HBB:c.46delT.
Xmn1 polymorphism |
Beta thalassaemia major |
Hb E beta thalassaemia |
|---|---|---|
Xmn1++ |
0 |
17(16.35%) |
Xmn1-/+ |
5(4.81%) |
55(52.88%) |
Xmn1-/- |
14(13.46%) |
13(12.50%) |
Total |
19(18.27%) |
85(81.73%) |
The most common type of mutation detected in patients was Cd 26 (G-A) + IVS 1-5(G-C) seen in 58 patients, out of which 39(67.24%) were heterozygous for Xmn1 polymorphism, 8 (13.79%) were homozygous for Xmn1 polymorphism and 11(18.97%) ) were negative for Xmn1 polymorphism.
Xmn1 polymorphisms |
-/- |
-/+ |
+/+ |
Total |
|---|---|---|---|---|
Cd 26(G-A)+IVS 1-5(G-C) |
11(18.97%) |
39(67.24%) |
8(13.79%) |
58 |
Cd26+30(G-C) |
1(7.14%) |
5(35.71%) |
8(57.14%) |
14 |
Cd26+130(G-C) |
0 |
3(100%) |
0 |
3 |
Cd26+15(G-A) |
1(16.66%) |
4(66.66%) |
1(16.66%) |
6 |
Cd26+Fr42-43 |
0 |
1(100%) |
0 |
1 |
Cd26+Fr 8-9) |
0 |
2(100%) |
0 |
2 |
Cd 26+(15-T) |
0 |
1(100%) |
0 |
1 |
IVS 1-5(G-C)+30(G-A) |
2(66.67%) |
1(33.33%) |
0 |
3 |
IVS 1-5(G-C) +Fr 8-9) |
1(100%) |
0 |
0 |
1 |
IVS 1-5(G-C)+15(G-A) |
2(40%) |
3(60%) |
0 |
5 |
IVS 1-5(G-C) +IVS 1-5(G-C) |
9(90%) |
1(10%) |
0 |
10 |
Total |
27 |
60 |
17 |
104 |
The second most common mutation detected was Cd 26+30(G-C) detected in 14 patients where 8 (57.14%) patients were homozygous (+/+), 5(35.71%) were heterozygous and one (7.14%) was negative for Xmn1 polymorphism. In thalassemia major patients with mutation IVS1-5(G- C)+IVS1-5 (G-C), 9(90%) out of 10 cases were negative for Xmn1 polymorphism and one (10%) was heterozygous for Xmn1 polymorphism (table III).
Diagnosis |
Xmn 1 - allele |
Xmn 1 + allele |
|---|---|---|
Betat thalassaemia major(no19) |
33(0.868) |
5(0.132) |
Hb E beta thalassaemia(no 85) |
81(0.476) |
89(0.524) |
Total |
114(0.548) |
94(0.452) |
Discussion
This is the first study of the frequency of Xmn1 polymorphism in Bangladeshi thalassaemia patients and its association with thalassemia mutations.
A total of 104 thalassaemia patients were studied where beta thalassaemia major was 19(18.27%) and Hb E beta thalassaemia were 85(81.73%). There were 57 males (54.81%) and 47(45.19%) females. The mean age at the time of diagnosis in our study was 13.04 years in Xmn1+/+ patients, 7.91 years in Xmn1 +/- and 5.50 years in Xmn1 -/-.
The presence of Xmn1 polymorphism in both heterozygous and homozygous state has an influence in later presentation of the disease as reported in some studies. Panigrah et al reported later presentation of Hb E beta thalassaemia patients in the presence of heterozygous Xmn1 polymorphism.22 Sharma et al reported in his study of thalassaemia major patients, the mean age of presentation was 18.3 months with homozygous Xmn1 +/+ and less than one year in Xmn1-/-.23 Ali et al found the mean age of thalassaemia patients with Xmn1 polymorphism was 8 years+7.9 while those without Xmn1 was 5.8+4.5.24
In Hb E beta thalassaemia patients in our study, heterozygous Xmn1 polymorphism (-/+) was seen in 55(52.88%), homozygous (+/+) in 17(16.35%) and 13(12.50%) patients were Xmn1 (-/-). In a study in China on major Hb E beta thalassaemia, the frequency of heterozygous polymorphism Xmn1 -/+ was 65.6% with no homozygous Xmn1 +/+ or Xmn1 -/-.25
In Malaysia, a study on Chinese and Malays thalassemia patients, where most of the patients were Hb E beta thalassaemia, there was marked variation in Xmn1 detection between Chinese and Malays. In their study they found homozygosity for the Xmn1(-/- was higher in Chinese beta thalassemia major patients (89.7%) while in Malays it was 28.6%. Homozygosity for Xmn 1(+/+) was absent in Chinese patients while it was 8.2% in Malaysian Malays.26
In Eastern India, a study in thalassaemia patients where all the patients were Hb E beta thalassemia and of the 128 chromosomes studied, 62 detected Xmn1 polymorphism while 66 were negative for Xmn1 polymorphism and the ratio of + allele to – allele was 0.94.The most common genotype found was heterozygous Xmn1 polymorphism (-/+) detected in 79.6% 27 In our study also, a majority of patients were Hb E beta thalaessemia and 81 chromosomes were -/- and 89 detected Xmn 1 polymorphism, and the ratio of + allele to - allele was 1.09.The most common genotype in our study was heterozygous Xmn1(-/+) 57.70%.
In Thalassaemia major patients in our study, Xmn1 polymorphism were -/- in 14(13.46%) cases and heterozygosity (-/+) in 5 (4.81%) patients and there was no homozygosity +/+ for Xmn1 polymorphism. Low positivity of Xmn1 polymorphism has been reported in thalassaemia major patients in Egypt (3.5%, 4%), Pakistan (3.3%) and Turkey7.4%.16,17,28,29
The mutant allele frequency of Xmn1 polymorphism was 0.45 and most of our patients were Hb E beta thalassaemia which was closer to a report in Eastern India where the allele frequency was 0.48 and all patients were also Hb E beta thalassaemia.27 It was lower in comparison to a study in Malaysia where the allele frequency was 0.66 and the majority of patients were also Hb E beta thalassaemia.26 Allele frequency varied in different countries and different regions of the same country. In Malaysia 0.66,in eastern India 0.48,northern India 0.27,western India 0.25,in western Iran 0.39 and 0.41 in Southern Iran.26,27,30-33.
The most common beta thalassaemia mutation detected was IVS 1-5(G-C) and the second common mutation was 30(G-C). IVS-1-5(G-C) in combination with Cd26(Hb E beta thalassaemia) were seen in 58 patients. Here 39(67.24%) patients were heterozygous (-/+) for Xmn1 polymorphism and 8 (13.79%) were homozygous (+/+), and in 11 patients (18.97%) no Xmn1 were detected. The next common beta thalassaemia mutation detected was 30(G-C) seen in 14 patients in combination with Cd 26.(Hb E beta thalasssaemia). Here also 8 (57.14%) patients were homozygous for Xmn1 polymorphism and 5 (35.71%) were heterozygous with only one patient (7.14%) did not have Xmn1 polymorphism. Verma also reported very high percentage (84.8%) of presence of Xmn1 polymorphism in B0-HbE thalassemia patients suggesting that Xmn1 polymorphism may be linked to Hb E chromosome.21 This is substantiated by reports of other studies including ours.25,27
The association of certain beta thalassaemia mutations with Xmn1 polymorphism has been reported in some studies. In Moroccan thalassemia patients, presence of Xmn1 polymorphism was found to be linked to the mutations Fsc(6-A),codon 24 (A-T) and 256bp deletion.20 In Dezili Turkey,Bahadir et al observed that Xmn1 polymorphism was relatively higher in Bo IVS-11-1, Bo codon 44 and B+-87 mutations in comparison to other B thalassaemia mutations.17 In Pakistan, Xmn1 polymorphism association with Beta thalassaemia varied in different studies. Ali et al observed IVS1-5/IVS 1-5 to be associated with Xmn1 polymorphism.24 Ashraf et al with IVS-1-1.34 and Hanif et al with IVS1-5,Cd30,IVS II-1and Inv /Del Gã(A ãä â).35 and in Egypt Said et al reported higher IVSII - 1 mutation in Beta thalassaemia major patients.28 Correlation was also seen between Xmn1 polymorphism and IVS-1 mutation in the Punjab Khatri community in India.19 In Iran Xmn1 polymorphism was detected in homozygous state in 87.5% of patients for the IVS-II-1 (G’!A) mutation.18
Therapy with Hydroxyurea has also been shown in many studies to reduce the requirements of blood transfusion or completely no blood transfusion in thalassaemia patients. An Iranian study reported favorable response to hydroxyurea in thalassaemia major patients where 80% had good response in patients having Xmn1 homozygous and 40% in heterozygous Xmn1.36 A study in Israel on the response to hydroxyurea to thalassaemia major and intermedia patients resulted in majority of thalassaemia major patients became transfusion independent and those thalassemia intermedia patients requiring occasional transfusion did not require any further transfusion. Here, majority of thalassaemia major patients were either homozygous or heterozygous for Xmn1 polymorphism.37
A study in India also showed good long term response to hydroxyurea therapy in patients with Xmn1(+/+). Other genetic factors like Beta thalassaemia mutations, alpha thalassaemia mutations or polymorphisms in BCL 11A and HBS1L –MYB genes did not contribute to this response.38
Ansari et al in Pakistan reported good response with hydroxyurea therapy in a majority of Beta thalassaemia patients with Xmn1 polymorphism.11On the other hand, the response to hydroxyurea has not been very encouraging in some studies. Ehsani et al did not find any significant haematological response in presence of Xmn1 polymorphism in thalassaemia intermedia patients..14 Dixit et al reported good response to hydroxyurea in thalassaemia intermedia patients but it was not related to either Beta thalassaemia mutations or presence of Xmn1 polymorphism.13
Conclusion
The positivity of Xmn1 is quite high in our patients mainly in Hb E beta thalassaemia patients and its association with Cd 26 + IVS 1-5(G-C) and Cd 26 + Cd 30(G-C). Physicians may follow the presentation of these patients with these mutations and use hydroxyurea to reduce or eliminate the requirement of blood transfusion. A clinical study assessing the response to hydroxyurea of thalassaemia patients showing positive Xmn 1 polymorphism should be done.
Acknowledgments
The authors are thankful to Bangladesh Medical Research Council for financial support to conduct this study and also thankful to all thalassaemic patients\who agreed to give samples for this study.
References
- Khan WA, Banu B, Amin SK, et al. Prevalence of Beta thalassemia trait and Hb E trait in Bangladeshi school children and health burden of thalassemia in our population. DS (Child) HJ. 2005; 21:1-6.
- Angastiniotis M,Eleftheriou A,Galanello R ,Harteveld CL,Petrou M,Traeger-Synodinos J,Giordano P,Jauniaux E,Modell B,Serour G,Prevention of Thalassaemias and other Haemoglobin Disorders: Volume 1: Principles[Internet]. Old J, editor.2nd ed.Nicosia(Cyprus): Thalassaemia International Federation:2013.
PMID: 24572827 - Mannan A, Kawser J, Ahmed AMA, et al. A Demographic Approach for Understanding the Prevalence of Thalassemia Patterns and Other Hemoglobinopathies: Selective Study in Chittagong City Perspective. Asian Journal of Biological Sciences. 2013;6:124-30.
- Uddin MM, Akteruzzaman S, Rahman T,et al.”Pattern of â- Thalassemia and Other Haemoglobinopathies: A Cross- Sectional Study in Bangladesh.” ISRN Hematol. 2012; 2012: 659191.
- Khan WA, Banu B, Sadiya S, Sarwardi G (2017). Spectrum of types of thalassemias and hemoglobinopathies: study in a tertiary level children hospital in Bangladesh. Thalassemia Reports. 7:6354
- Italia K, Jain D, Gattani S, et al . Hydroxyurea in sickle cell disease - A study of clinico-pharmalogical efficacy in the Indian haplotype. Blood Cells Mol. Dis. 2009;42:25-31.
- Olivieri NF, Weatherall DJ. The therapeutic reactivation of fetal haemoglobin. Human Molo. Genet. 1998; 7:1655-1658.
- Winichagoon P, Fucharoen S, Chen P, Wasi P. Genetic factors affecting clinical severity in beta thalassemia syndromes. J Pediatr Hematol Oncol . 2000; 22:573–580.
- Thein SL. Genetic insights into the clinical diversity of â thalassaemia. Br J Haematol. 2004; 124:264-74.
- Dedoussis GV, Mandilara GD, Boussiv M, Loutradis A. HbF production in â-thalassaemia heterozygotes for the IVSII-1 G-A â0-globin mutation. Implication of the haplotype and the (G)gamma) “158 C-T mutation on the HbF level. Am J Hematol. 2000; 64:151-56.
- Ansari SH, Shamsi TS, Munzir S,et al.Gã-Xmn 1 polymorphism: a significant determinant of â-thalassemia treatment without blood transfusion. J Pediatr Hematol Oncol. 2013; 35:153-56.
- Motovali-Bashi M, Ghasemi T. Role of XmnIG Polymorphism in Hydroxyurea Treatment and Fetal Hemoglobin Level at Isfahanian Intermediate â-Thalassemia Patients. Iran Biomed J. 2015 ; 19:177–82.
- Dixit A, Chatterjee TC, Mishra P,et al. Hydroxyurea in thalassemia intermedia—a promising therapy. Ann Hematol. 2005; 84: 441-46.
- Ehsani MA, Hedayati-Asl AA, Bagheri A,et al.Hydroxyurea –induced hematological response in transfusion independent beta-intermedia thalassemia: Case Series and Review of Literature. Pediatric Hematology and Oncology.2009;26: 560-65.
- Nuntakarn L, Fucharoen S, Fucharoen G,et al. Molecular, hematological and clinical aspects of thalassemia major and thalassemia intermedia associated with Hb E-beta- thalassemia in Northeast Thailand. Blood Cells Mol Dis. 2009;42:32-35.
- Tantawy AA, Andrawes NG, Ismaeil A,et al. Prevalence of Xmnl Gã polymorphism in Egyptian patients with â-thalassemia major. Annals of Saudi Medicine. 2012; 32:487-91.
- Bahadir A, Atalay EO. Frequency of GG-globin promoter -158 (C>T) Xmn I polymorphism in Denizli, Turkey. International Journal of Physical Sciences. 2012; 7:1927-31.
- Neishabury M, Azarkeivan A, Najmabadi H. Frequency of positive Xmn1 Gã polymorphism and coinheritance of common alpha thalassemia mutations do not show statistically significant difference between thalassemia major and intermedia cases with homozygous IVSII-1 mutations. Blood Cells Mol Dis. 2010; 44: 9-9
- Kaddah N, Risk S, Kaddah AM,et al.Study of possible genetic factors determining the clinical picture of thalassaemia intermedia. Journal of Medical Sciences. 2009; 9: 151-155.
- Agouti C, Badens A, Abouyoub M,et al.Genotypic correlation between six common â-thalassemia mutations and the Xmn1 polymorphism in the Moroccan population. Hemoglobin. 2007; 31: 141–49.
- Verma IC, Kleanthous M, Saxena R,et al.Multicenter study of the molecular basis of thalassemia Intermedia in different ethnic populations, Hemoglobin. 2007;31:439-52.
- Panigrahi I, Agarwal S, Gupta T, et al.Hemoglobin Eâ thalassemia: factors affecting phenotype. Indian Pediatrics. 2005; 42:357-62
- Sharma N, Das R, Kaur J, Ahluwalia J.et al. Evaluation of the genetic basis of phenotype heterogeneity in north Indian patients with thalassemia major.Eur J Hematol. 2010; 84:531-37
- Ali N, Ayyub M, Khan SA,et al. Frequency of Gã-globin promoter -158 (C>T) Xmn1 polymorphism in patients with homozygous/ compound heterozygous beta thalassaemia. Hematol Oncol Stem Cell Ther. 2015; 8:10-15.
- Liu RR, Wang MY, Lai YR. Analysis of Gã-158(C’!T) polymorphism in hemoglobin E/â-thalassemia major in Southern China. J Hematol Oncol. 2010; 3:29.
- Wong YC, George E, Tan KL, et al Molecular characterization and frequency of Ggamma Xmn I polymorphism in Chinese and Malay beta-thalassaemia patients in Malaysia. Malays J Pathol. 2006; 28:17-21.
- Bandyopadhyay S, Roychowdhury K, Chandra S, et al. Variable severity of -thalassemia patients of eastern India: effect of á-thalassemia and xmnI polymorphism. Clin Exp Med. 2001; 1: 155-59.
- Said F, Salam AA. Xmn1 polymorphism: Relation to B – thalassemia phenotype and genotype in Egyptian children. Egyptian Journal of Medical Human Genetics.2015;16: 123-27
- Hanif TB, Ahmed S, Anwar J, et al. Xmn1 polymorphism and disease severity in patients with Beta Thalassemia major from Northern Pakistan. J Ayub Med Coll Abbotabad. 2015;37:12-16
- Kumar R, Kaur A, Agarwal S. Influence of Xmn 1Gã (HBG2 c.-211 C ’! T) Globin Gene Polymorphism on Phenotype of Thalassemia Patients of North India. Indian J Hematol Blood Transfus. 2014 30: 286–90.
- Nadkarni A, Gorakshakar AC, Lu CY, et al. Molecular pathogenesis and clinical variability of beta-thalassemia syndromes among Indians. Am J Hematol. 2001; 68:75–80.
- Hooshang N, Zohreh R, Gholamreza B. The Xmn1 polymorphic site 5’ to the Gã-gene and its correlation to the Gã: Aã ratio, age at first blood transfusion and clinical features in â-thalassaemia patients from Western Iran. Mol Biol Rep. 2010; 37:159-64
- Karimi M, Yarmohammadi H, Farjadian S,et al. Beta- thalassemia intermedia from southern Iran: IVS-II-1 (G>A) is the prevalent thalassemia intermedia allele. Hemoglobin. 2002 ;26:147-54.
- Ashraf S, Moinuddin M. Inheritance of IVSI-I Mutation Assures 158 (C-T) Xmn1 Polymorphism in Thalassemia Intermedia. International Journal of Hematology and Oncology. 2015;4:230-35.
- Hanif TB, Ahmed S, Anwar J, Kazmi SK. Xmn1 Polymorphism and disease severity in patients with beta thalassaemia from Northern Pakistan. Journal of Ayub Medical College, Abbottabad : JAMC. 2015 ; 27:13-16.
- Yavarian M, Karimi M, Bakker E,et al .Response to hydroxurea treatment in Iranian transfusion dependent thalassaemia patients. Haematologica. 2004; 89:1172-78
- Koren A, Levin C, Dgany O,et al. Response to hydroxyurea therapy in beta-thalassemia. Am J Hematol. 2008; 83:366-370.
- Italia K, Chandrakala S, Nadkarni A.et al.The Genetic Determinants for Long-term Response to Hydroxyurea Therapy in Indian â-Thalassemia Patients. Clin Res Hematol. 2019; 2:1-8.

Submission
25 February 2021
Accepted
30 June 2021
Published
01 August 2021
Apply citation style format of Bangladesh Medical Research Council
Issue
Vol 47 No 2 (2021)
Section
Research Articles
Ethical Clearance
NREC of Bangladesh Medical Research Council (BMRC), Dhaka, Bangladesh.
Financial Support
Bangladesh Medical Research Council (BMRC), Dhaka, Bangladesh.
Conflict of Interest
There was no conflict of interest.